高载量原子分散催化剂制备
当金属颗粒降低到一定尺度(纳米尺寸甚至原子级分散)时,由于其较高的原子利用效率和独特的电子特性,负载型金属催化剂往往会展现出极高的催化活性和特定的选择性。而且原子级分散催化剂具有特定的配位活性中心,便于从分子层面理解催化机理,是沟通多相催化与均相催化的桥梁。然而,随着金属颗粒尺寸的降低,金属的表面自由能会急剧增大,很容易导致金属团聚。传统的解决方案通常以牺牲金属载量(< 2 wt%)来制备原子分散催化剂,这极大地限制了该类催化剂的实际应用。
中国科学技术大学梁海伟教授课题组与林岳老师以及中国科学院高能物理研究所储胜启副研究员合作,使用高比表面积硫掺杂介孔碳(meso_S-C)为载体,基于贵金属与硫之间的强键合作用原理,制备出一系列高载量原子级分散贵金属催化剂,包括Pt、Ir、Rh、Ru以及Pd,其中金属Pt的载量可以达到10 wt% (如图1)。该研究成果发表在《Science Advances》上(Sci. Adv. 2019, 5: eaax6322)。在该工作中,研究人员首先利用他们前期发展的过渡金属催化碳化有机小分子的方法(Nature Communications 2015, 6, 7992;Science Advances 2018, 4, eaat0788),合成了具有高硫含量(~14 wt%)和高比表面积(>1200 m2 g-1)的meso_S-C载体,并利用传统的浸渍法在该载体上成功制备出5种原子级分散的贵金属催化剂。
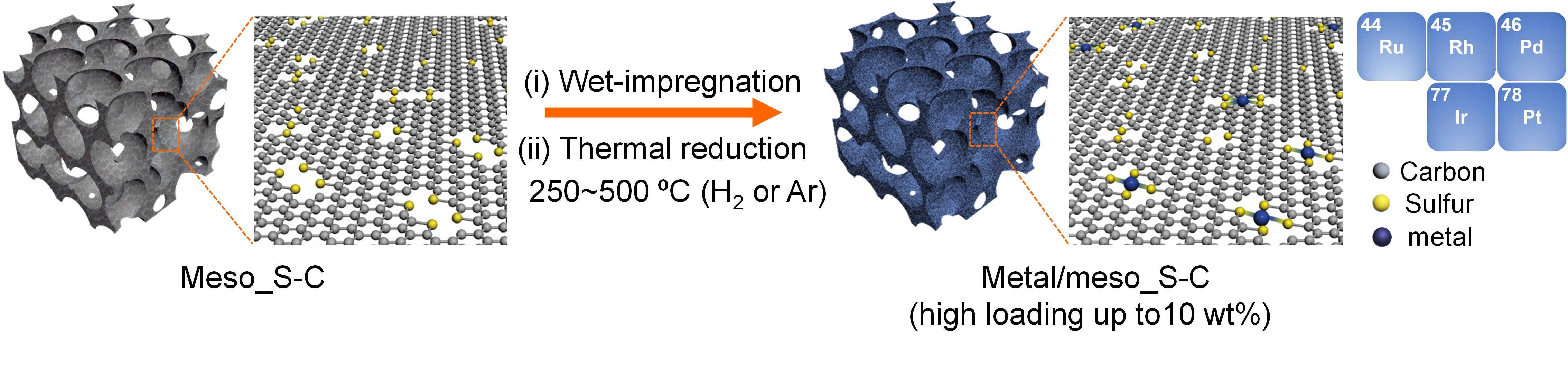
图1 原子分散贵金属催化剂的合成示意图。
从球差校正透射电镜图来看,当金属Pt载量小于10%时未发现纳米颗粒或纳米团簇存在(图2A-B),而在20Pt/meso_S-C和30Pt/meso_S-C(图2C-D)催化剂中很明显观察到团簇或纳米颗粒。X-射线吸收精细结构谱(XAFS)是表征原子分散催化剂的最强有力的手段之一。如果样品中金属以原子形式分散在载体表面就不会有金属-金属键存在,反之样品中若有金属团簇或颗粒存在就能检测到金属-金属键。研究人员依托北京同步辐射装置1W1B-XAFS实验站获得的X射线近边吸收精细结构(XANES)表明,Pt/meso_S-C中的Pt处于氧化态(图2F)。扩展X射线吸收精细结构(EXAFS)结果表明(图2E),10Pt/meso_S-C,在2.5-2.8埃范围内没有出现Pt-Pt键的峰,而在1.8-2.0埃处很明显观察到Pt和其他元素的配位键。与PtO2相比,该峰的位置要比Pt-O键更长,因此该峰可归属为Pt-S键。拟合结果表明,当Pt载量小于10wt%时,只有Pt-S键存在(配位数CN=3.6),没有Pt-Pt键,证实了Pt催化剂以原子分散的形式分散在meso_S-C上,同时验证了金属与硫的键合作用。随着Pt金属载量的提高,Pt-S配位数降低,同时Pt-Pt键的配位数增加。此外,随着Pt含量的升高,Pt-Pt键的键长逐渐升高(从2.717埃到2.763埃),且比Pt片中Pt-Pt键的键长(2.763埃)短,说明Pt原子从无序到有序变化这一过程。
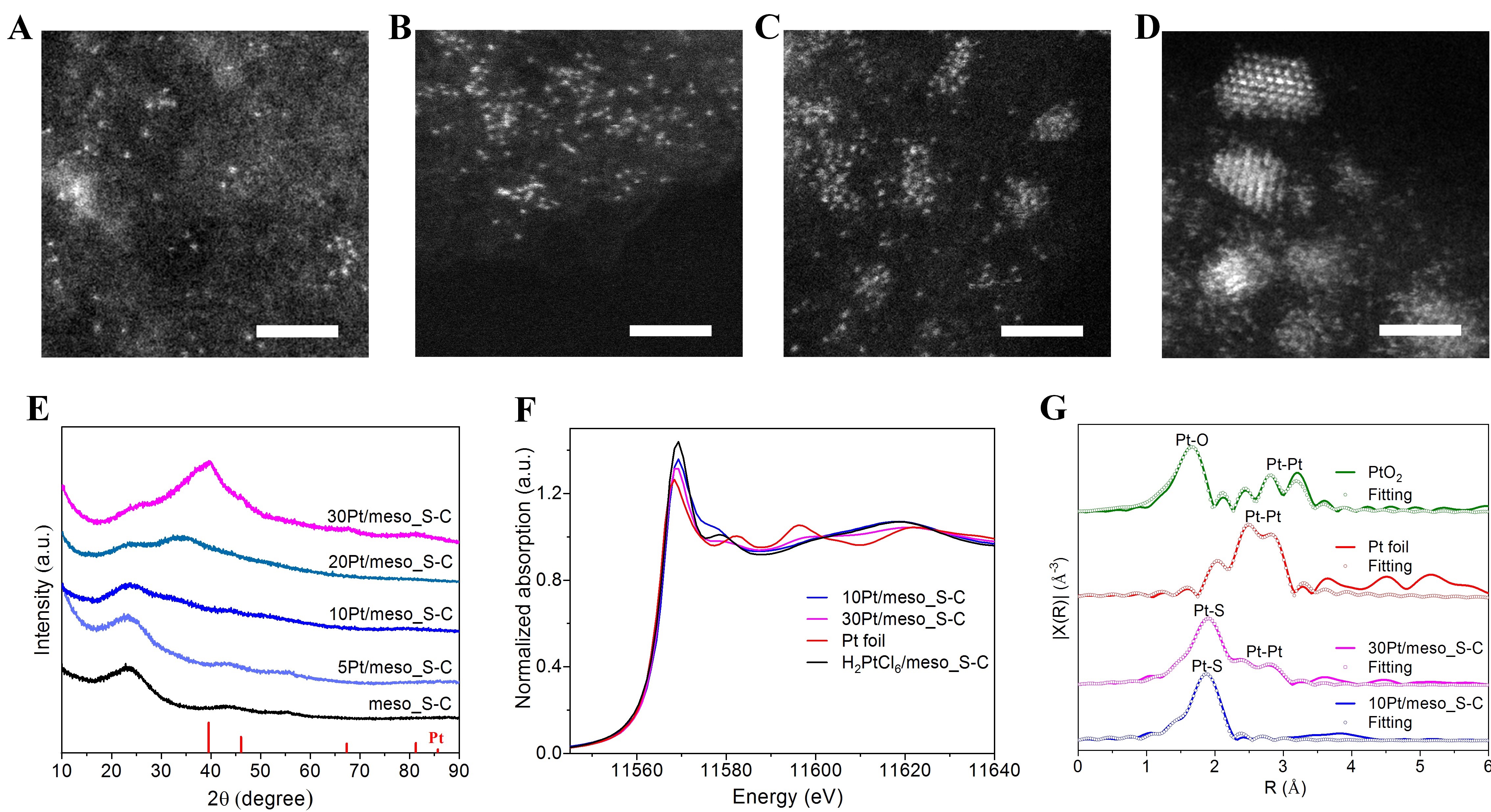
图2 Pt/meso_S-C催化剂表征。
同时在meso_S-C载体上还制备了高载量原子级分散的Ir、Pd、Rh和Ru催化剂,使用球差电镜未发现没有明显的金属原子团聚(图3A-D),使用XAFS进行表征,同样在2.5-2.8埃范围内没有出现金属-金属键的峰,而在大于金属-氧键的位置检测到金属-硫的配位(1.8-2.0埃),表明这些贵金属催化剂以原子形式分散到meso_S-C上。
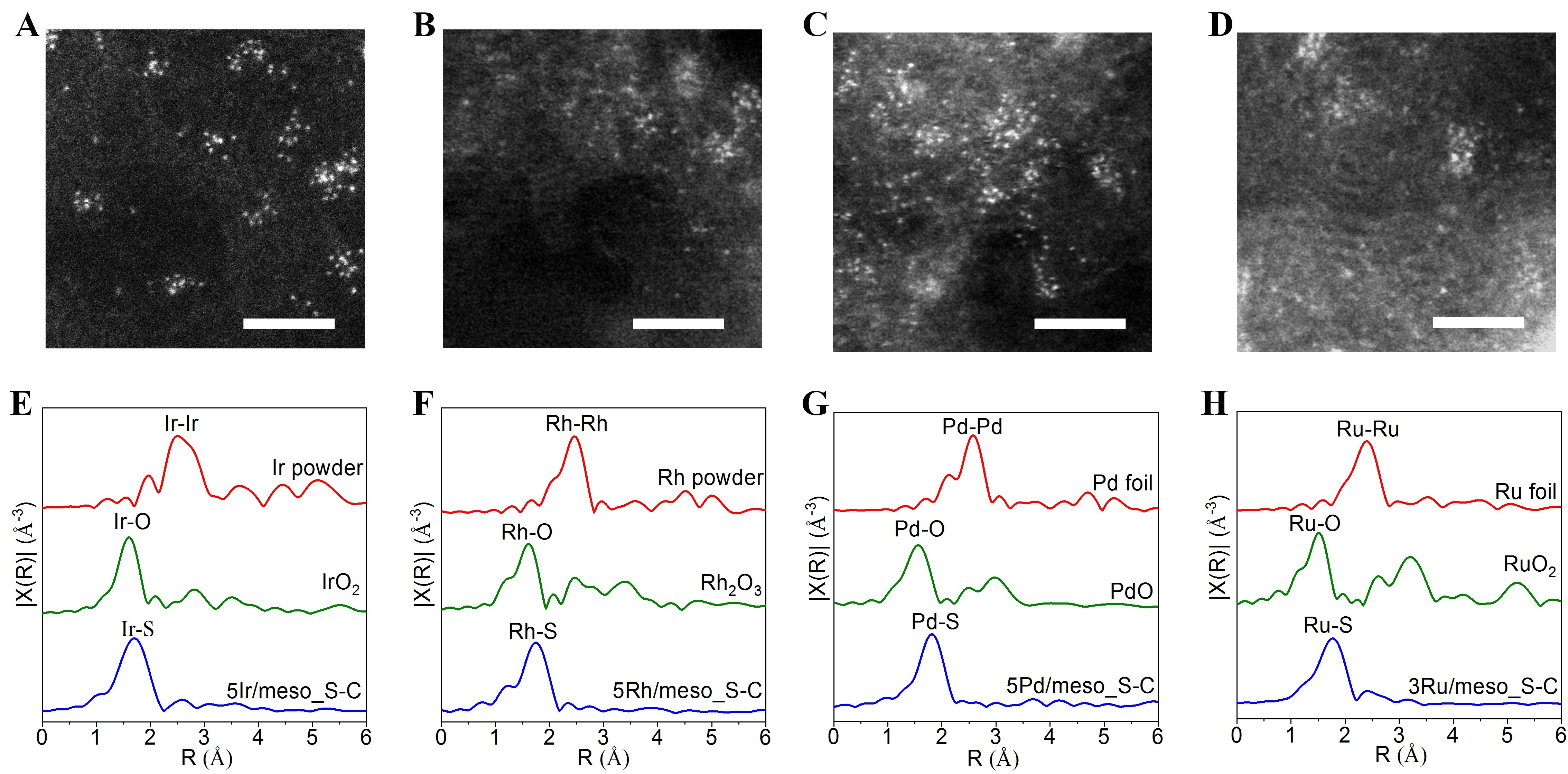
图3 原子级分散催化剂的电镜和EXAFS表征。
在电催化甲酸氧化反应中,原子级分散Pt催化剂(10Pt/meso_S-C)质量活性达到2.38埃,是商业Pt/C催化剂的30多倍。在催化喹啉加氢反应中,原子级分散Ir催化剂(5Ir/meso_S-C)的TOF值达到1292h-1,是商业Ir/C催化剂的20多倍。该项工作为高载量原子级分散贵金属催化剂的普适性制备提供了一种新的思路。
发表文章:
Lei Wang, Ming-Xi Chen, Qiang-Qiang Yan, Shi-Long Xu, Sheng-Qi Chu*, Ping Chen, Yue Lin*, Hai-Wei Liang*. A sulfur-tethering synthesis strategy toward high-loading atomically dispersed noble metal catalysts. Sci. Adv. 2019, 5: eaax6322.